safrole
beilstein/crossfire search
2002, by 3basezip
search as: substance, product, reactant
recomended browsers: mozilla, explorer
browsers who doesn't support the bmp format
won't display the reaction pictures.
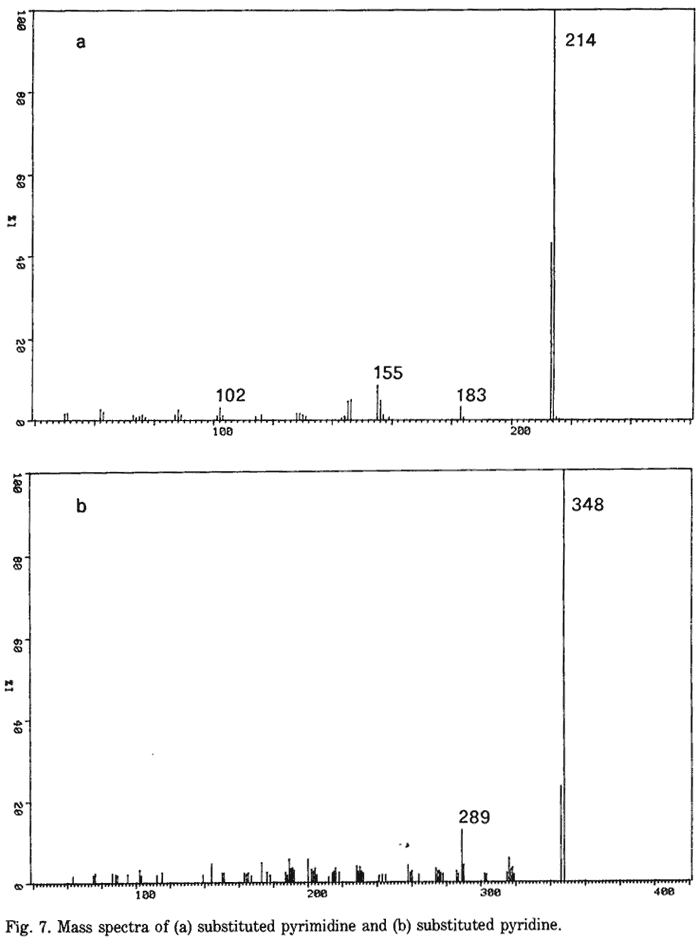
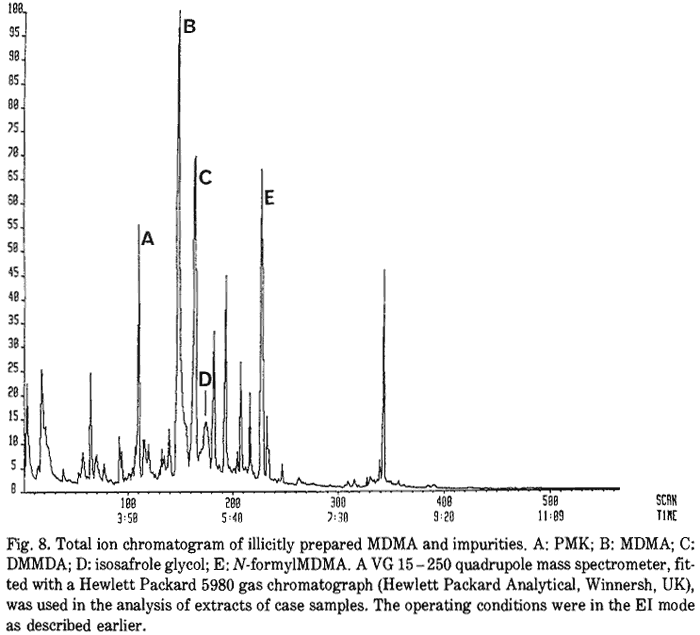
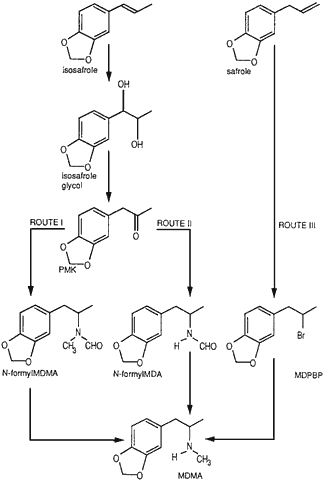 Fig. 1.
Fig. 1.
Compound
|
Relative retention time
|
(MDMA = 1.0)
| |
Safrole
|
0.35
|
trans-Isosafrole
|
0.44
|
cis-Isosafrole
|
0.53
|
PMK
|
0.97
|
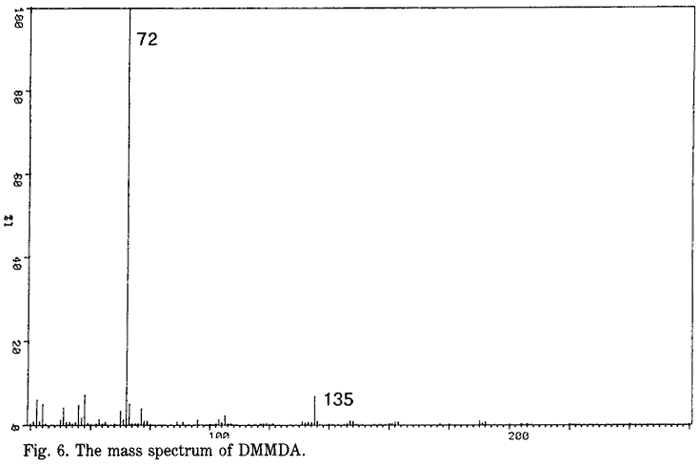
DMMDA
|
1.21
|
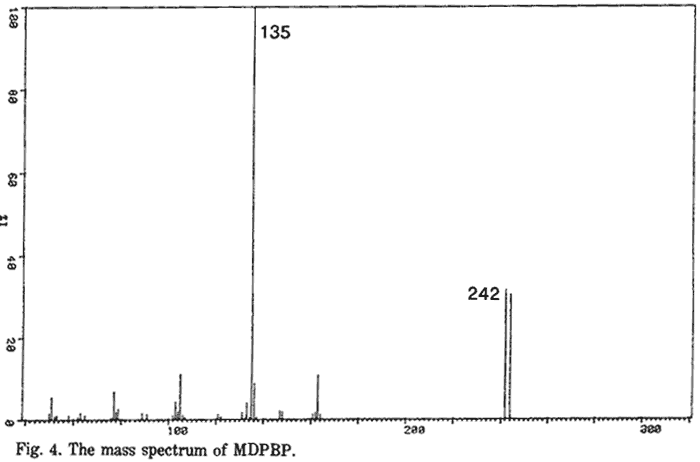
MDPBP
|
1.44
|
Isosafrole glycol
|
2.24
|
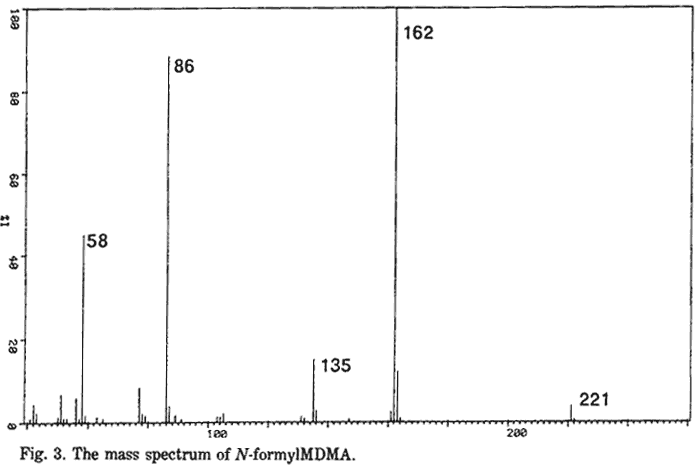
N-formyl-MDA
|
5.77
|
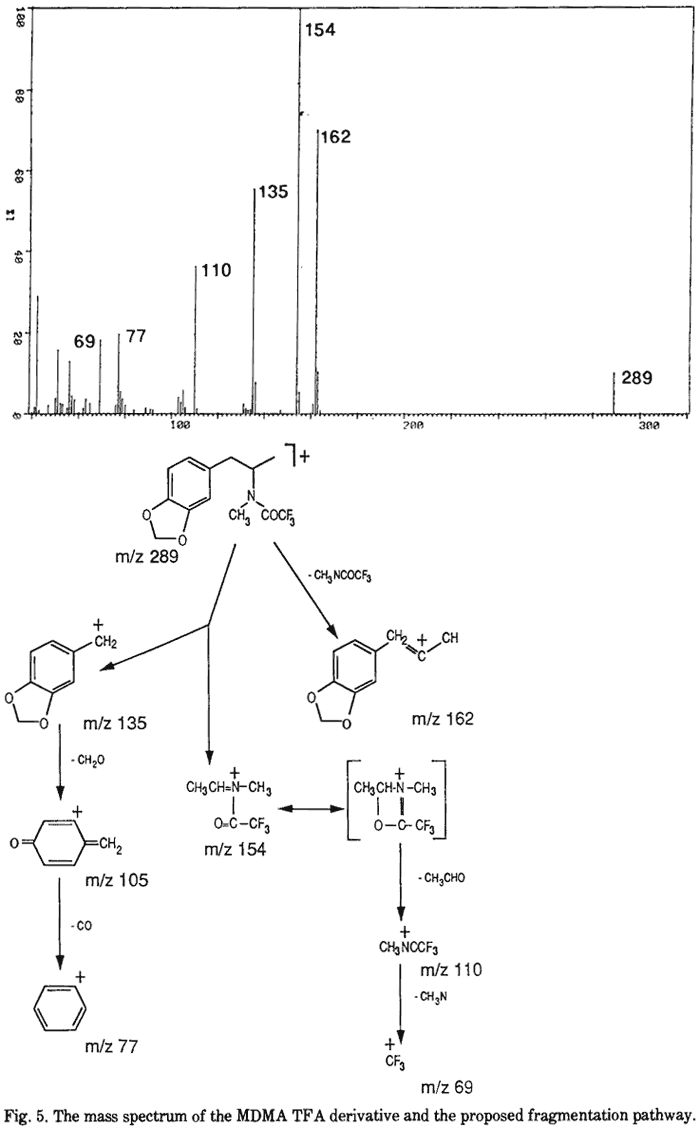
N-formyl-MDMA
|
6.45
|
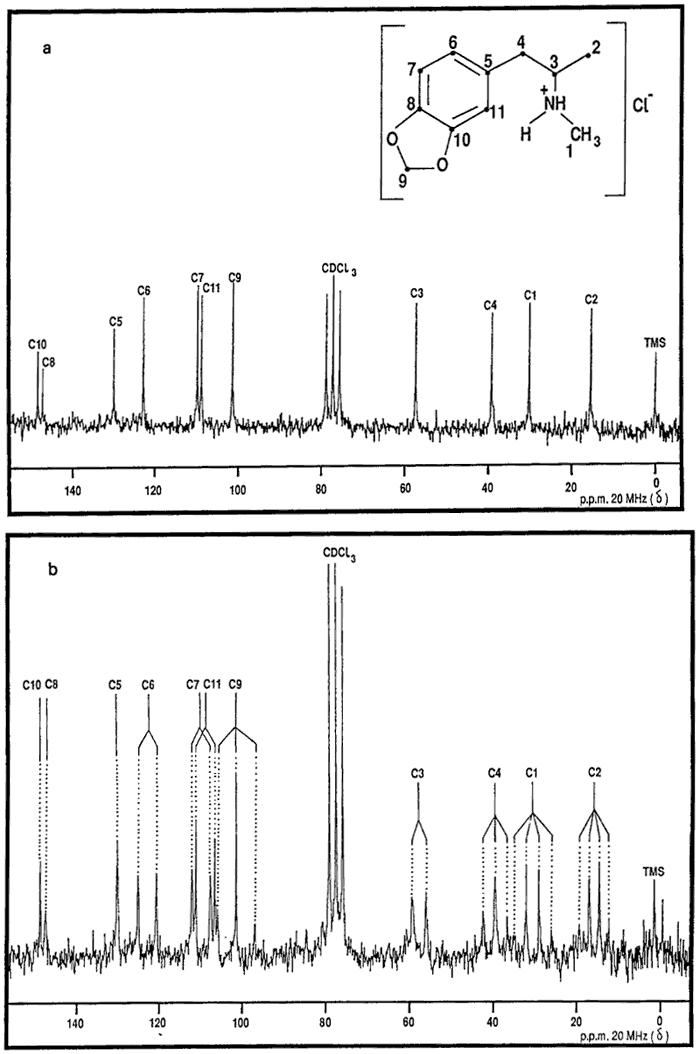
Chemical shift
(BBPD, Fig. 2a) |
Multiplicity
(SFORD, Fig. 2b) |
Assignment
|
15.4
|
quartet
|
C-2
|
30.2
|
quartet
|
C-1
|
39.1
|
triplet
|
C-4
|
57.3
|
doublet
|
C-3
|
101.2
|
triplet
|
C-9
|
108.7
|
doublet
|
C-11
|
109.6
|
doublet
|
C-7
|
122.6
|
doublet
|
C-6
|
129.8
|
singlet
|
C-5
|
146.9
|
singlet
|
C-8
|
148.1
|
singlet
|
C-10
|
Compound
|
Relative Retention Value
|
MDMA
|
0.27
|
DMMDA
|
0.38
|
N-formyl-MDA
|
0.75
|
N-formyl-MDMA
|
0.78
|
Peak
| Compound Name |
MW.
|
Formula
|
A
| Safrole |
162
|
C10H10O2
|
A
| Isosafrole |
162
|
C10H10O2
|
B
| Piperonal |
150
|
C8H6O3
|
C
| 4-Allyl-1,2-dimethoxybenzene |
178
|
C11H14O2
|
D
| Piperonylmethylketone (PMK, MDP2P - Main Component) |
178
|
C10H10O3
|
E
| 3,4-Methylenedioxyphenyl- 2-propanone-(3-ol) |
194
|
C10H10O4
|
F
| 3,4-Methylenedioxyphenyl- 1-propanone |
178
|
C10H10O3
|
G
| 3,4-Methylenedioxyphenyl- 1-butanone |
172
(sic) |
C11H12O3
|
H
| 4-Isopropyl-1,6-dimethyl- 1,2,3,4-tetrahydronaphtalene |
202
|
C15H22
|
I
| 3,4-Methylenedioxyphenyl- propionic acid-2-one |
208
|
C10H8O5
|

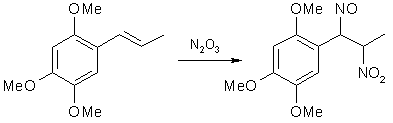 Fig 1. Formation of Asarone Pseudonitrosite
Fig 1. Formation of Asarone Pseudonitrosite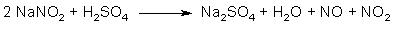 Fig 2. N2O3 from NaNO2 and H2SO4
Fig 2. N2O3 from NaNO2 and H2SO4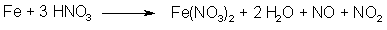 Fig 3. N2O3 from HNO3 and iron
Fig 3. N2O3 from HNO3 and iron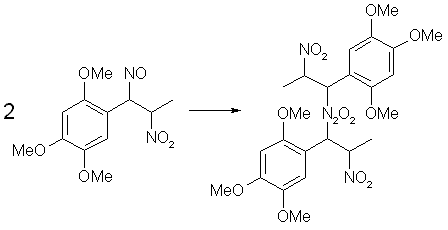 Fig 4. Dimerization of Asarone Pseudonitrosite
Fig 4. Dimerization of Asarone Pseudonitrosite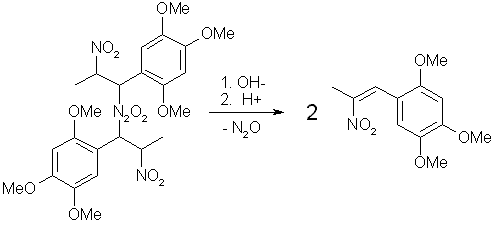 Fig 5. Hydrolysis of Asarone Pseudonitrosite Dimer
Fig 5. Hydrolysis of Asarone Pseudonitrosite Dimer| Pseudonitrosite | Color | Yield | Melting point |
| Asarone | Yellow | 16.5g, 58% (22.8g/80%)4 | 130°C(dec)4 |
| Isosafrole | White | 77% (18.4g)5 | 128°C3 |
| Anethole | White | 48% (10.8g)7 | 126°C(dec)7 |
| Nitropropene | Color | Yield | Melting point |
| Asarone | Yellow/red* | 19.7g, 78% |
101°C4
|
| Isosafrole | Yellow |
98°C5
| |
| Anethole | Yellow |
47°C7
|
Photoamination of MethylstyrenesThe Possible One-pot-shot From Isosafrole to MDMAby Drone #342By far, one of the most fascinating possible methods of making MDMA is the insanely elegant photolytic reaction between isosafrole and methylamine. For years, like many other individuals, I looked at isosafrole and methylamine and dreamed that somewhere there was a method of adding the two together in a single step, rather than having to make MDP-2-P, then reductively aminate it, and go through all the tedious steps involved. As it turns out, such a method exists: photo-induced nucleophilic amination. The general idea is this: an alkoxy-substituted methylstyrenes (in this case, isosafrole), p-dicyanobenzne (DCB), methylamine, and triphenylbenzne (TPB), are disolved in acetonitrile, exposed to light, and the reaction occurs. Here's the mechanism: p-DCB absorbs a photon, gets activated, and abstracts an electron from isosafrole's double bond, leaving the carbon beta to the benzene ring (in the 2-position) positively charged. From there, the nucleophile (in this case methylamine) attacks the aforementioned carbon, and releases a hydrogen. The hydrogen is then scooped up by the cationicly-charge alpha carbon. The basis of this technology is some research done in 1973, involving the addition of MeOH across double bonds. During the early 90's, some Japanese researchers had the ingenuity to apply this to ammonia as a nucleophile, then tried alkylamines as well. Both were greatly successful, and were directly applied to the synthesis of phenethylamines. Fortunately for MDMA enthusiaists, several papers of theirs focused on the synthesis of methoxylated aryl isopropylamines -- which is exactly what MDMA is. Though MDMA was never expressly synthesized by these fellows, one can easily apply the techniques they developed to MDMA production. General method for the production of amphetamines from methyl styrenes, lifted from the literature."General procedure of Photoamination. Into a Pyrex vessel was introduced an MeCN-H2O (9:1, 70 mL) solution containing 1-4, 6-8 [various styrenes] (3.5 mmol) and DCB (3.5 mmol), and then the solution was bubbled with gaseous ammonia. The solution was irradiated by an eikosha PIH-300 high pressure mercury vapor lamp under cooling with water. The progress of the reaction was followed by GLC analysis..." Proposed experimental method.Proposed procedure of photomethylamination of isosafrole. Into a Pyrex vessel was introduced an MeCN-H2O (9:1, 70 mL) solution containing isosafrole (C10H10O2; 0.567 grams, 3.5 mmol) and DCB (3.5 mmol). The solution was then saturated with methylamine gas. The solution was irradiated by a 300-W high pressure mercury vapor lamp under cooling with water. Upon completion, the solvent was stripped in vacuo, and the residue was washed an aqueous 10% NaOH solution, and extracted with DCM. The DCM was stripped in vacuo, and the oil distilled in vacuo, yielding the free base of MDMA. The base was disolved in dry Et2O, and dry HCl gas was bubbled in. The precipitated crystals then dried, weighed, and tested for biological activity.By definition, this is an oxidative amination. For old MDMA chemhacks, familiar with reductive amination, this should sound a little weird, but hey, it works. Bibliography: a reading list of the good stuffNeunteufel, R. A., Journal of the American Chemical Society, 1973, 95, 4080-4081This is the original study of photo-induced nucleophilic addition that inspired the later use of ammonia. While its only marginally useful, it's a good place to start to understand some of the theory of this process. Tetrahedron Letters, 1993, 34, 5131-5134This is a quick, general sneak-preview of the later study of phenethylamine synthesis. Nice, basic, but not too much detail Tetrahedron, 1994, vol. 50, no. 31, 9275-9286"Photoinduced Nucleophilic Addition of Ammonia and Alkylamines to methoxy-Substituted Styrene Derivatives" This is the Mack-daddy of all the articles, IMHO. Here's where they show what they got, and lay out the procedure and conditions that would be used for making MDMA. Bulletin of the Chemical Society of Japan, 1998, vol 71, no 7, 1655-1660"Photoamination of Alkenylnaphthalenes with Ammonia via Electron Transfer" A further study, where aryl isopropylamines are made from their corresponding alkenes. This one's good for several reasons: the use of methylamine, and the evidense it has proving the usefulness of both isomers of isosafrole. The Tetrahedron article by the same research group before used only the trans isomer, but not the cis. Links to other selected photochemistry (and otherwise related) web pages.US Pat #4,483,757:Photochemical process for preparing aminesUS Pat #4,459,191: Light-catalyzed process for preparing amines |

The following is quoted from: Journal of Organic Chemistry 53, 4081-4 (1988)Thats amazing! NINETY FOUR freakin percent from an aqueous MeAmine soln!!! Try achieving that with a halogen leaving group on our favorite alkene by cooking the stinky shit up in a pipe bomb w/ alcoholic MeAmine. You can't, if you are lucky 50% will be . Halogens blow as leaving groups compared to sulfonate derivatives as proved by this paper. It is such an advantage to be able to use good 'ol fashioned aqueous MeAmine compared to homebrew anhydrous alcoholic amine solns! The only drawback noticed immediately is the large excess of MeAmine employed by the authors. This shouldn't present a huge problem because the excess amine cooked off during the heating process can be collected by bubbling through HCl. The 65°C rctn temp is the bp of THF so excess amine will be liberated out of the top of the reflux condensor on rctn pot. A slow stream of N2 can be admitted through a bubbler in the second neck of the rctn. flask forcing the methylamine gas to be expelled out the top of the condenser into a beaker of HCl. Simply evap off HCl to obtain your excess amine back as MeAmine hydrochloride.
(R)-Tomoxetine Hydrochloride: A solution of phenyl-3-(2-methylphenoxy)-propyl methane sulfonate [the mesylate group is on the gamma carbon] (450mg, 1.45mmol) and methylamine (10ml, 40% in water) in THF (10ml) was heated to 65'C for 3h. After cooling, the solution was diluted with ether, washed w/ aq. sat. NaHCO3 soln. and brine, and dried with anhydrous K2CO3. After concentration a pale yellow oil was obtained which was dissolved in ether, bubbled w/ dry HCl gas [and you guys certainly know the rest] to produce a white ppt which was recrystallized w/ acetonitrile to yield title compound (400mg, 94%)
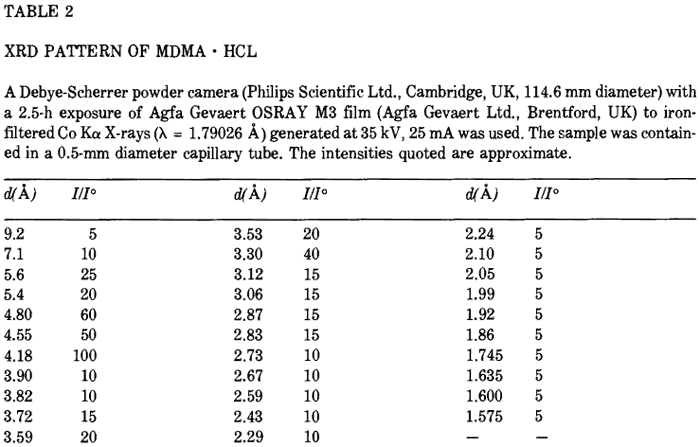{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}