MDMA Synthesis Story
What follows is, of course, a work of pure fiction. In it, I have tried to illustrate something of the mental state of an individual whose mind was severely altered when he took his first dose of ecstasy and decided that he would love to take more. Doctor Drool, the protagonist, was paranoid about taking street drugs, but was quite confident of his own abilities in a chemistry lab, although he had not exercised those abilities for 20 years. He was moderately well trained in chemistry in the university (two full years of inorganic and organic classes with labs), but he had not done or even thought about lab work for many years.
The manufacture of ecstasy is, of course, totally illegal and degenerate, and the author feels that anyone who tries to do it should be hauled in front of John Ashcroft's military tribunals and then executed (or executed first; it doesn't really matter).
Although in the story that follows, the protagonist has so far avoided arrest and execution, I'm certain that he will receive the punishment he deserves any day now.
Any relationship between the methods described by Doctor Drool and reality are, of course, completely coincidental, but to make the story more believable, I have included text from actual web pages, perhaps written by real degenerates, not like the fictional Doctor Drool.
I hope you enjoy the story. If it keeps just one person from falling into the hell of ecstasy (or, as the addicts sometimes call it, "rolling" on MDMA, ADAM, E, X, or XTC), it will have been worth the effort of writing.
-- the author, Feb 25, 2002
Introduction:
I initially tried to follow
the BrightStar synthesis that I found on the internet but the only part that was reasonable was for the distillation of safrole from the raw sassafras oil. It was, however, good to read BrightStar's description just to remind myself about some chemistry techniques that I hadn't done for 20 years. I later found other syntheses on the internet for all the steps between legal chemicals and MDMA that worked quite well.
At least at the current time (early 2002), all the chemicals and apparatus mentioned here are available legally. Aside from the ecstasy, of course, the only other intermediate products that you will produce along the way that are at the least highly suspicious are the pure, distilled safrole and the ketone MDP-2-P. The unfortunate thing about the ketone is that although you probably have a million good places to hide the ecstasy and the safrole, the MDP-2-P must be kept in the freezer. The folks from the DEA know this. Luckily, the ketone is only necessary for the final synthesis, so I made it just before I needed it, and then repeated the final step until the ketone was all used up. Now when the DEA breaks down my door, they've got to find less than an ounce of white powder (maybe I should label it "anthrax") in an unknown form rather than find a little jar of antifreeze-colored green fluid in my freezer.
Make certain that you have (or know you can obtain) all of the stuff (chemicals and apparatus) before you begin. For example, at the time BrightStar wrote his synthesis, it may have been easy to obtain dimethylformamide; now, it is not. If you decide to embark upon this project, it is not going to be cheap, assuming you're starting from scratch. I spent perhaps $1100 total -- $800 for lab supplies (glassware, hotplate-stirrer, clamps, stands, vacuum pumps, et cetera), and $300 for chemicals. If I decide to do it again, it'll probably cost me another $200 (and I'll get a lot more product the second time).
My overall impression is that the synthesis is moderately difficult, at least for someone who last took an organic chemistry lab course more than 20 years ago. I was quite good in the labs back then, and also understood the chemistry quite well. I took the time to understand to a pretty good degree the chemistry in this synthesis. The more anal-compulsive you are, the better you'll do. Study the reactions. Understand how they work. Although it would seem that you could just follow a cookbook method, it's amazing how much better that works when you know exactly what you are doing and why you are doing it.
If I had tried this synthesis without experience with organic chemistry laboratory techniques, I have severe doubts that I'd have been able to pull it off. If you are without experience but would still like to try, remember that most junior colleges offer lab courses in organic chemistry. Take one of those and concentrate on getting an A+ in the lab part.
Overall notes:
(This stuff is in no particular order -- there may be more important stuff following less important stuff.)
This process is going to take a long time. Even if you had all the stuff you needed, it takes a few days. There are lots of long waits for reactions to proceed, and you'll find you need to go shopping for things you forgot. Also, what I tended to do is when I got a particular step to work, I'd rerun that reaction a bunch of times in a row to make a lot of the product for future steps. If you run a reaction the next day, you'll remember everything about it. And you will make mistakes in the later steps, destroying some of your intermediate product, so it's nice to have made enough to be able to recover and try again right away. The BrightStar synthesis led me to believe that the entire process might just take a couple of long days. I think it took me a month between purchasing the first chemical and "rolling" for the first time on my own stuff. I hope these notes will reduce your time significantly.
You can check yourself as you go along. For example, if the boiling points of your intermediate products are wildly out of line, you've screwed up. Note the expected colors, consistency, et cetera.
One of the referenced documents on the web contains photos from the final production of MDMA from the ketone and nitromethane. They don't look very appetizing (other than the final photo of beautiful, brilliant, white crystals), but it's great to know that your gunk looks exactly like his gunk. You're going to eat the stuff later, right? Almost all the precursors are poisonous, right? You really would prefer to make something "edible", wouldn't you?
The scariest part, perhaps, is your first test of your first final product, unless you happen to have a mass spectrometer or an NMR in your basement (I don't). Luckily, I had tasted real MDMA previously, so I could taste the tiniest grain of my final product. Even though MDMA tastes pretty shitty, when my stuff tasted the same as the real stuff, it was the best-tasting thing in the world! Then, if you've got good sense, try a little bit for your first real dose -- perhaps a quarter of what you normally take (100 or 125 mg is a "normal" dose, depending on your weight, susceptibility,
et cetera). Then try a half dose, then the whole thing.
I have seen "test kits" available that are presumably used to test street drugs to make certain that they're what they were sold as. I think that at least most of them are not highly specific tests; they simply indicate that the chemicals are in some general class. This might be good enough to tell you that the MDMA you bought is really speed or LSD in place of ecstasy, but if you've made the stuff yourself, it's pretty unlikely that you'll have speed or LSD in your final product by accident. Your precursors are all MDMA precursors, so the test kit is far more likely to "see" MDMA-like chemicals than other stuff, even if you totally screwed up. So it probably wouldn't hurt to run one of these tests, but it won't tell you for sure, and it certainly won't give you any indication of the purity.
I am completely paranoid, so, except for the totally safe reactions (and the way you'll know which ones are safe is by studying the reactions), I kept a fire-extinguisher handy. The only reaction that scared the shit out of me was the distillation of the nitromethane from the RC fuel. I did it outdoors, and for that one had the pin pulled on the fire extinguisher all the time.
Also, wear old clothes, with long sleeves, long pants, shoes and socks, and you might want to use rubber gloves when you're working with the highly corrosive stuff like muriatic acid, sulfuric acid, and sodium hydroxide. Safety glasses (or at least regular glasses, if you wear them) are a good idea for many steps. Although I thought I was being careful, I was not highly amused after working with the concentrated NaOH solution to find about a hundred tiny holes in my pants after washing them. I apparently splattered them with NaOH, and it ate a bunch of little holes...
I spent perhaps $300 on chemicals and, including a whole bunch of errors, made about 60 grams of pretty pure ecstasy. That's 600 doses of 100 mg. At $20 per dose, that's $12,000 worth. If you scrimp on the chemicals, perhaps you'll spend only $200, but a likely result is that you will then get $0 worth of ecstasy. The $100 savings nets you a $12,000 loss. I'm not much of a businessman, but that seems like a bad deal to me...
Get the right stuff if you can. Buy real dichloromethane (methylene chloride); don't mess with the Jasco stripper, since it's got alcohol and God knows what else in it. Use distilled water -- you can get it for a couple of bucks at the grocery store (or make it yourself, since you've got distillation glassware anyway). Get technical grade methanol, xylene, et cetera. You may spend an extra $100, but, again, that's just 5 doses of ecstasy. I used stuff like baking soda (NaHCO
3), Salt (NaCl), epsom salts (MgSO
4·7H
2O),
et cetera, right from the grocery store or drugstore.
You can get a lot of the chemicals from mail order, Ebay, et cetera, but you will probably have to face some humans to buy stuff that's slightly suspicious. Don't volunteer stories of why you need them for your kid's science fair experiment -- just say nothing. It's good to have in mind some legal uses, but they won't ask. Don't get everything at once for two reasons. First, a complete grocery list for MDMA precursors is pretty suspicious, and second, the DEA in the US requires that large (over $100) purchases of chemistry supplies be reported to them. I got little bits everywhere, and made three trips to the lab supply place to get all my precursors (only two trips would have been necessary to keep the bills under $100 each, but, par for the course, I fucked up).
The main reason to go to a lab supply place is for the stuff that it's a pain to mail-order because of shipping constraints. Things like xylene are highly flammable, so it's much easier to get a gallon of that at the lab place than to hassle with the shipping. Similarly, muriatic acid is easy to get from a pool supply store, and get the nitromethane (mixed with methanol and castor oil) at a hobby shop.
Here's an idea I didn't use but you might: At the lab supply place, ask for the safety sheets on one or two of the chemicals. They're required to have them, and for some reason people think that if you're at all interested in safety then you're not the total degenerate that you really are.
Even when you're not at the lab supply, it feels "safer" if you know the lingo. At the pool supply place where you get the hydrochloric acid, don't ask for "hydrochloric acid"; ask for "muriatic acid". That's what pool people call it. God knows why. Similarly, in the hardware store, ask for "DampRid", not "anhydrous calcium chloride".
I know it feels like the time you were 16 years old and went into the drugstore to get "some chewing gum, some lifesavers, ..., oh, and a package of condoms", but you'll get over it.
Have enough ice on hand for the job. Again, ice is relatively cheap, so get an extra bag for each session. It's a royal pain in the ass to run out in the middle of some critical process. In addition to running ice water through the condenser for distillations, I also used ice in the bucket I used to recycle water through the aspirator. Ice water makes for a better vacuum, and the better the vacuum, the better your vacuum distillations will work. Remember that even when you are just using ice to cool a reflux column, you sometimes need to cool it for up to eight hours.
Work in a ventilated area or under a fume hood (since none of us have fume hoods in our houses, that means a ventilated area), especially when you're working with the stuff like the xylene, acetone, and especially with the HCl gas. In fact, do those outside if you can (definitely outside with the HCl). Look -- you're planning on causing enough brain damage as it is with the ecstasy; why add to it with xylene brain damage that doesn't even make you feel good?
Be patient -- for example, remember that every couple of drops of MDP-2-P that you toss away is a dose (perhaps a $20 street value) of ecstasy. When you're doing the separations, wait for the stuff to settle out properly, or you're throwing good stuff away. While you're waiting for a separation, I'm sure there's some glassware you can clean! And do use very clean glassware!
Get silicone grease to seal the distillation joints during vacuum distills. Make certain it's well sealed. If you turn on the pump and it leaks, turn off the pump, re-seal, and restart. If you don't, you're throwing away good stuff (or possibly toasting it, which is far worse). Brightstar, for example, recommends vaseline, instead of the silicone grease but I figure why not shell out a couple of bucks more and get
exactly what works best.
If you screw up, don't continue without correcting the problem. Don't just assume that some of that brown gunk is what you wanted in spite of the fact that the instructions here say that you should be getting a bunch of brilliant white crystals. Before you start a process, be sure you know exactly how to do it. Read an organic lab techniques book on the subject -- it's amazing what you can learn and how much you can retain when you're sufficiently motivated.
If you are the least unsure, do a dry run before you commit the real chemicals. For example, just put water into the system and go through the motions the first time. I don't know what you'll learn, but I'm sure you'll learn something. I did a distillation of eugenol from oil of cloves just to make sure I knew how to do a vacuum distillation of an oil with a high boiling point before I committed my precious sassafras oil. You can get oil of cloves easily from a drugstore, and it's easy to look up boiling points of eugenol, et cetera. Similarly, if you've never used a separatory funnel, pour in some water and oil, shake it up, and then separate those. You'll learn to control the valve, you'll learn whether the valve needs greasing with silicone grease, you'll learn about venting, et cetera.
Be sure to get a good collection of jars that are easy to pour from. It's a giant pain to try to pour fluid from a jar and have it dribble all over your hands. (Guess how I learned this?) The Pyrex measuring cups are great for pouring. Some jars are; some aren't. Check them by pouring water from them before you put in stuff that you care about.
Beware of breaking the plug on the separatory funnel. If you're staring at the liquid and don't have your hand on the plug and the pressure builds up, it's certain to pop out when you're carrying it over a concrete floor. (Guess who broke his?) In fact, the glassware is all moderately delicate, so try to do anything you can to avoid putting it at risk. If you really are just beginning, hook up your various glassware arrangements over a soft bed the first time. Make sure stuff is securely clamped. Pay attention!
Read everything you can before you start. I read and re-read the entire synthesis many times, and then before I started any single step, I re-read it again, and kept a copy next to me as I worked. Notice that there are plenty of things you can do ahead of time -- make aluminum foil balls, "cook" MgSO
4·7H
2O (epsom salts) to make an anhydrous powder, et cetera.
Don't be in a hurry, especially for the last couple of steps. If you screw them up, you waste ALL your work. If you're going to be sloppy, do it at the beginning so you don't waste too much of your effort :^)
After I got the first working batch, I confess that I did the second batch in a "modified" state of mind, and it worked fine. I was able to keep everything in mind and under control, and, if anything, was able to concentrate better on what I was doing.
Stuff you'll need:
(I may easily have left something off this list; be sure to read the individual parts of the synthesis.) The recommended amounts below allow for a bunch of mistakes. Believe me; you'll make them. I started with a quart of sassafras oil, just to give you the scale, and used about 2/3 of it.
Chemicals:
- Sassafras Oil: this is about 80-90% safrole. Look for aromatherapy supplies. You can get it on the web. Be sure to get 100% sassafras oil -- don't get it mixed with anything else. It's about $45 per quart, and a quart is vastly more than you'll need, but I'd get at least a half quart (I guess that's a pint).
- MeOH (methanol, wood alcohol): I got mine from a lab supply house, technical grade. I'd get a gallon or two to account for fuck-ups. Maybe you can get it at drug stores, but all I've seen there is denatured ethanol and isopropyl alcohol. I eventually used two gallons.
- Distilled H2O: Distill it yourself, or get it at the grocery store. You'll need a few gallons (perhaps 5 or 6) by the time you're done. Get distilled water, not "spring water" or some other such shit.
- p-Benzoquinone: Get this from photography supply shops. I got a pound and had plenty for a lot of trials.
- Palladium(II)chloride (PdCl2): Photography supply shops. This is expensive stuff ($25/gram). I got 5 grams total. Get exactly this -- not a replacement or a mixture of this and something else. You can do it 1 gram at a time, but the "recipe" below calls for 2. If you only have 2 grams, do two 1-gram batches.
- DCM (methylene chloride; dichloromethane): I got it from a chemical lab supply house, technical grade. I used less than a gallon.
- Xylene: Chemical supply house. I used technical grade, and eventually went through an entire gallon.
- Acetone: It's with the paint thinners in a hardware store. I got a gallon and it was more than enough. Mostly, I used it to wash and dry glassware, but you also need a little bit for the final wash of the sacred chemical.
- NaCl (non-iodized table salt): grocery store. Be certain it's not iodized. I don't know what the iodine would do, but I have a healthy imagination, and none of the things I could imagine were good. I think almost all salt (maybe not kosher salt) contains sodium aluminum silicate. Mine did, and that didn't seem to cause any problems. It's cheap -- get three pounds.
- NaHCO3 -- Sodium bicarbonate (baking soda): grocery store. One big box is plenty.
- NaOH (lye): I used Red Devil lye from a grocery store (drain cleaner). Get pure, dry lye. Not Drano with the little aluminum flecks, or anything that's already dissolved. Get three or four cans of the stuff.
- HCl (hydrochloric acid, muriatic acid): Get this from a pool supply place. I got a gallon, and it was far more than enough. It's cheap. Get a gallon or two.
- MgSO4·7H2O -- Magnesium sulfate: These are epsom salts. Get them at a drugstore. You'll need to cook it to get rid of that "·7H2O". I bought the amount that fits in a container that's the size of a half-gallon milk carton.
- Nitro RC fuel: From a hobby shop that sells fuel for the RC models. I got a gallon for around $30, and it was far more than I needed. Get the highest concentration you can. The best I found was (supposedly) 40%. It's a mixture of nitromethane, methanol, and something like castor oil. I hear that even higher concentrations are available. The kid that sold me the 40% stuff seemed really concerned that I'd ruin my model airplane engine using such a high concentration. I assured him that I'd dilute it with some methanol -- yeah, right.
- Aluminum foil: I used the "extra heavy duty" Reynolds wrap. One roll from the grocery store is plenty. Apparently the stuff called "heavy duty" also works, but I assume the reaction will run faster (and hotter and scarier) with it. Everybody cautions against the regular foil. It is just too thin, and believe me, you don't want to see that reaction "run away". It barrels along at a pretty heart-stopping rate even with the extra heavy duty stuff.
- Peanut oil: Grocery store. Get a couple of quarts.
- Safflower oil: Grocery store. A little bit; all you need is a few ounces. Be sure to get the stuff with NO ADDITIVES since it's going to get hot and be mixed with something you eventually eat!
- HgCl2 (mercuric chloride): From a photo chemical supply place. You don't need much -- 3 or 4 grams is plenty. I think I got an ounce and used only a tiny amount. Maybe there's some way I can feed the rest to certain politicians. Careful: this stuff is REALLY poisonous.
- Sulfuric Acid: I used the high-powered drain cleaner. I got it from a hardware store. I used less than a quart. I don't need to say "be careful", do I?
- CaCl2 (Calcium chloride): I used "Damp-Rid" that I got from a hardware store. It's with the mildew prevention chemicals. One cannister is plenty.
Apparatus:
Be sure it all fits together. I used 24/40 connectors throughout. The 24/40 seems to be the most easily available stuff, and it seems quite suitable for the quantities of chemicals you'll be using. If you already happen to have larger or smaller stuff and just need a piece or two, go ahead and use that, but if you're starting from scratch, get the 24/40 size.
If you haven't done this sort of thing for awhile (or ever), obtain a manual on organic lab techniques. Read through the appropriate section before you begin using any technique. Of course you'll screw up anyway, but this way you'll screw up a couple of fewer times. The manual recommended by many illicit chemists is this:
"The Organic Chem Lab Survival Manual ", by
James W. Zubrick. Others work fine, too.
For me, at least, it was critical to have something like the "CRC Handbook of Chemistry and Physics" or the "Merck Index" to look up physical properties (especially boiling points) of such things as methanol, acetone, safrole, eugenol, ...) It gave me a warm, fuzzy feeling when the stuff would distill at the correct temperatures, and when there was a problem, I'd know about it.
You can find lots of this apparatus on the internet, and can sometimes get good prices on places like Ebay on used glassware.
Glassware:
- 1000 ml 2-neck round-bottomed flask (RBF)
- 500 ml RBF
- distillation head
- vacuum adapter
- thermometer coupling
- vigreaux column
- distillation column
- separatory funnel: at least 1000 ml -- 2000 or 4000 ml is better.
- addition funnel: at least 100 ml
- 2000 ml two-neck RBF (not required, but nice)
(If you're lucky, you may be able to find a separatory funnel with a 24/40 fitting at the end that you can use as an addition funnel as well. I was not lucky, so became the proud owner of yet another piece of glassware.)
Other Stuff:
- Buchner filter, filter flask, flask stopper, filter paper.
- Coffee filter + filter paper
- Combination hot plate/stirrer. You can't get away without this, especially for the final step, and it makes life a lot easier if you have it at the beginning. I got mine late and was sorry.
- Magnetic stir bar (get one that's egg-shaped and works in RBFs)
- Clips to hold the glassware together.
- Two lab stands and at least two clamps. Don't skimp on the clamps -- they hold hundreds of dollars worth of glass!
- Aquarium pump (to pump ice water through distillation column)
- Thermometer that fits coupling (0-300 degrees). It's nice to have two -- one to watch the temperature of the stuff that's coming over in the still, and another to check other temperatures, like the temperature of the peanut oil, or of the fluid that you are distilling.
- Boiling stones. You can often avoid using these if you've got the hotplate/stirrer. The spinning stir-bar works better than boiling stones under vacuum conditions, since there tends to be a lot less "bumping". If you've never seen bumping, let me tell you a secret: the first time it happens to you, you'll be very upset.
- Silicone grease
- Vacuum pump. I used a spa pump with enough pipes to take water out of and return it to a 5 gallon plastic bucket.
I attached an aspirator to this.
- Plastic tubing (3 pieces -- two to connect to the input and output of the distillation column; one to connect the aspirator to the vacuum adapter, or to the flask holding the buchner funnel). Make sure they're long enough. I used 2 sections that were about 2.5 feet and one of 6 feet.
- Measuring cups and/or graduated cylinders. I used a 500 ml cylinder plus a 50 ml cylinder.
- Triple-beam balance.
- Glass wool (I got a furnace filter and cut out chunks of the wool). I used it to plug the vigreux column when it was filled with glass beads and calcium chloride).
- Lots of clean glass jars for storage. Next time I'd just get a dozen mason jars (quart size) with lids. Some smaller jars are good, too.
- Tiny spatula for measuring out tiny amounts of dry chemicals.
- Glassware cleaning brushes. Get at least one curved one for cleaning the insides of the round flasks, and one straight one.
- Glass beads to fill the vigreux column for fractional distillations. The people at the hippie store that sold all sorts of psychedelic colored beads thought it was pretty strange when I looked over everything and finally selected the ugliest (and of course cheapest) beads for my "creation".
- Coffee grinder (to ball up the aluminum foil) Please clean out the coffee grounds first, and PLEASE clean out the aluminum dust afterwards.
- (optional) paper shredder (to shred aluminum foil)
Synthesis notes
(All temperatures below, unless mentioned otherwise, are centigrade.)
Overview:
- I distilled sassafras oil to make safrole.
- I distilled RC model fuel to make nitromethane.
- I used the Wacker reaction to make MDP-2-P from the safrole.
- I made MDMA from the MDP-2-P and the nitromethane.
- I took the ecstasy. This step was more fun than all the other steps put together!
Safrole:
There seems to be no problem with the distillation of the safrole from the raw sassafras oil using
Bright Star's recommendations.
If a ¾ horsepower pool pump is used, it appears that the temperature at which the safrole comes over is about 106-107°C on the first pass. For the second pass, I used a vigreux column in addition, and the safrole came over at between 94°C and 99°C. Good vacuum! The pump basically recirculates water through an aspirator as fast as it can using water in a 5 gallon plastic bucket. To get a higher vacuum, there was always ice in the bucket. Thus the temperature of the recirculating water was always 0°C (unless I ran out of ice).
There are a couple of photos on the web of
designs for good vacuum pumps that recirculate water. I mounted my pump to a board and then attached pieces of PVC pipe to make an output that sprayed into the bucket, and an input that sucked water from the bucket. I did not, therefore, need to drill any holes in the bucket as was proposed in at least one of the web designs.
The suction also seemed to be vastly better if there was a baffle in the plastic bucket to keep the air bubbles from getting sucked into the input of the pump. I used the lid of the bucket as a baffle and crammed it down vertically between the place where the water from the aspirator fixture sprayed in water/bubbles and the input pipe from the spa pump sucked it out. This uses ice very quickly -- without ice, it's amazing how fast a ¾ horsepower motor heats up 4 gallons of water. By the way, a ½ horsepower motor should be sufficient; I just happened to have one rated at ¾ horsepower.
Also, always run ice water through the distilling column, either when distilling or when you're using it as a reflux column.
Distilled the sassafras over peanut oil in a 1000ml flask in 2 passes. The result was about 700ml of safrole from 1 quart of sassafras oil.
Nitromethane:
Bought nitro fuel for RC engines at a model hobby shop. The bottle said it was 40% "nitro" (= nitromethane), but after my distillation, I doubt it. It seemed to be at most 30%. Had to do a fractional distillation using a vigreaux column. The fuel is a mixture of methanol, nitromethane, and castor oil.
Note by Rhodium: This discrepancy is probably due to the fact that methanol and nitromethane forms an azeotrope. First a 92:8 MeOH/MeNO2 mixture (bp 64.5°C) distills over (close to the boiling point of pure methanol, bp 64.7°C) and when all the methanol is gone, the temp shoots up to ~100°C, where pure nitromethane is collected.
Nitromethane boils at 101°C, so I had to do it over cooking (peanut) oil instead of water. It seemed to come over at about the right temperature, but it's OK to have a mix of the nitromethane and some methanol, since the only time the nitromethane is used is in a mixture that uses methanol as a solvent, and the total amount of methanol is unimportant, so some extra is OK. If there is some methanol, however, it may be important to add a bit of extra nitromethane/methanol to be sure to get enough nitromethane, since that's critical to making the final product. I used about 10% or 15% more of the mix for that final step and didn't have any problems.
What I did was to distill for a long time to get almost all of the methanol out, and then, when the temperature spiked up to near 100°C, I let that run for a few seconds, then switched to a new flask to get the nitromethane.
Did I mention that this distillation scared the shit out of me?
What frightened me about the nitromethane is that the Merck Index says that it has a "flashpoint" at 112°F. I don't know what a flashpoint is, but I didn't want to find out! Nitromethane is the "nitro" in "nitro-burning dragsters" -- you know, those drag-racing cars with all the flames coming out. Note that the flashpoint is far below the boiling point (about 101°C) of nitromethane.
Note by Rhodium: This is not something of concern. The flash point of a substance is simply the lowest temperature at which you can ignite a substance. Gasoline has a flash point at several tens of centigrades below zero for example. It does not mean that the compound will spontaneously ignite or detonate, just that you should avoid sparks or open flames (which is a good rule at all times in a lab).
I tried to distill with a vacuum, and it was very difficult with boiling stones, since there would be a huge "bump" as a huge chunk boiled at once, which reduced the pressure, which had to build up for another huge bump. I did not have my hotplate/stirrer at the time, so I couldn't tell exactly at what temperature it was boiling, and the temperature was all over the place. Next time (if there is a next time), I'll try the vacuum distill, but with the stir-bar twirling like a dervish, and I'll use a vigreux column crammed with glass beads for a good fractional distillation.
Note by Rhodium: Distillation of nitromethane at atmospherical pressure is not inherently dangerous as long as you aren't distilling it all to dryness. It is not practical to vacuum distill nitromethane, as there is too much loss of product down the drain (using an aspirator as vacuum) or into your pump oil.
MDP-2-P (the "ketone"):
Used
Methyl Man's "Benzoquinone Wacker Oxidation of Safrole in Methanol".
I tried the long-term stirring of the PdCl
2 in methanol for 6 or 7 hours before adding the water and the p-benzoquinone. You can get the p-benzoquinone and PdCl
2 from photography supply places. I mail-ordered mine from a Canadian lab (I thought this was great, too, since I'll bet the DEA has a harder time looking at Canadian lab records). PdCl
2 is expensive -- like $25 per gram.
I never needed to add heat -- just a slow addition and then a long wait afterwards (8 hours to be sure everything reacted). Stirred the whole time.
I used gravity filtration in a coffee filter to get rid of the hydroquinone, and then put the filter in a zip-lock bag and squeezed it to get out the last drops.
When I washed with the sodium bicarbonate, I had a lot of crud on the interface between the DCM and the methanol fractions, but it was workable. The saturated NaCl made a real mess -- emulsion-like that just wouldn't separate. I finally froze it to break it up. I only did one NaCl wash since it was such a pain. Maybe it would be better not to shake the mixture so hard in the separatory funnel. Or maybe it would be better to use distilled H
2O for the mixes. Or both. Next time, I'd use only distilled H
2O for every place I need water, and I wouldn't shake so hard on this wash.
I did distill in two steps -- DCM over water without vacuum, then the MDP-2-P under vacuum over oil. Did it with a stir-bar and it worked OK unless I lost vacuum. Stir bar works much better than boiling stones, especially under vacuum.
I had problems cleaning the RBF afterwards, due to some charred shit. I read later that the best way to clean this is to add some paint thinner (mineral spirits) and clean with that FIRST, before you try stuff like soap and water (and strong acids, strong bases,
et cetera). I do know the mineral spirits don't work so well afterwards...
MDMA:
Used
Methyl Man's "Reductive Amination of MDP2P with Al/Hg + Nitromethane".
The reason I used this approach is that I was totally unable to make the methylamine hydrochloride (methylammonium chloride, MA.HCl) by any method. I tried BrightStar's method, as well as about 4 other methods I found on the web. In all cases, I was able to make what were probably mixtures of MA.HCl and ammonium chloride, but I couldn't figure out how to assess the purity. I also didn't have any luck getting the stuff I needed to do a purification: absolute ethanol is difficult to obtain; in California, the best you can purchase at a liquor store is about 75%, so you're faced with a distillation even to get to 95%. Then you've got to get some anhydrous calcuim oxide, and I didn't know how to do that. I even made a bunch of chloroform to clean out other impurities (which you should do), and that was a pain in the butt and perhaps a bit dangerous, too.
The problem is that the ammonium chloride will cheerfully react by the same mechanism as the MA.HCl with the MDP-2-P, but it will make MDA in place of MDMA. I would not have minded a mixture of, say, 3% MDA and 97% MDMA, but I just didn't know what I'd get. Anyway, the method described below worked perfectly, and should make exactly 0% MDA and 100% MDMA.
At first I didn't have a 2 liter, 2 necked RBF, so I did half-batches in a 1 liter flask, and that worked great, except that it took, of course, twice as long. There was never a problem with run-away reactions, and I always got a happy result. The full version is a little more tempermental, left more aluminum junk, but worked fine, too. Do this reaction at the end of the day, and let the final stuff sit around all night to make sure the reaction is complete. If you try to do it after just, say, four hours, there will be some aluminum fragments that are still bubbling in the NaOH, and these are a real pain in the butt during the separations. Wait overnight and everything is cool...
I used not only heavy aluminum foil, but the "Extra Heavy Reynolds Wrap". You can just cut 3 inch strips from the roll and put them through a paper shredder (to make pieces that are 3 inches long and ¼ inch wide) -- this is much easier than cutting them by hand. You'll have to do some dicking around with the coffee grinder to figure out how much of the foil strips can go in at once and not jam the machine. Even with the paper shredder and coffee grinder, it takes surprisingly long to make enough little foil balls to run the reaction.
After the reaction, I poured the grey sludge into large (gallon-sized) bottles, and added the xylene (xylene works fine instead of toluene, by the way) to the bottle. Then I did all the shaking in there before pouring the stuff in a separatory funnel.
I tried the washes with tap water and there was always a lot of gunk. On my final pass, I used distilled H
2O in all steps and things were MUCH better. The moral: use distilled water,
don't use tap water!
When you are drying the epsom salts (do this well ahead of time), don't put too much in the oven at once. I put a thin layer on a sheet of aluminum foil and cooked it very hot (400°F) in an oven for an hour or so. I made a bunch of batches and kept them sealed tightly in a jar. I also ground the stuff to a powder using a mortar and pestle. I might try a coffee grinder next time.
Don't try to cook up too much at once -- this can make a horrible mess.
I used a hair-dryer to dry the bottles that were to contain the anhydrous stuff (after the acetone wash, of course).
I had two bad experiences with HCl generators before I figured out how to do it. This is the way: Add NaCl (lots, non-iodized) to a two-necked flask. A big one (2 liters) is good. Wet the NaCl with HCl, but not too much (no puddles).
Plug a vigreaux column with glass wool (cut from a heater filter), and pour the column full of CaCl
2 (Damp-Rid pellets). Then put the thermometer adapter with a tube instead of a thermometer mounted in it into the top of the column.
Into the other neck of the main flask, put in an addition funnel with concentrated sulfuric acid. All joints should be greased with silicone lubricant. Drip in the H
2SO
4, and anhydrous HCl gas comes out the tube. Do this outdoors -- you're going to get clouds of HCl gas, which you certainly don't want to breathe. Use the silicone grease throughout.
Even at the end, when I thought there were no longer any mistakes to make, I screwed this up. I tried to powder the CaCl
2 to get better surface area, and then tried to pump through too much HCl gas too quickly. The powder was too fine, it plugged the gas, and finally a giant belch of gas, H
2O, and CaCl
2 was dumped into the xylene and it totally fucked up the batch. TAKE IT EASY AND SLOW WITH THE GAS GENERATION. You have plenty of time, and it's the last step.
TEST THE TUBE AND EVERYTHING IN XYLENE BEFORE YOU TRY IT FOR REAL! I found, to my horror, that the "aquarium rock" that I added to the end of the tube to make lots of tiny bubbles dissolves in xylene. I had great success with nothing but a hollow tube.
Don't be a pig -- when you get a bunch of precipitate, run it through the filter and then re-gas. It's even better to chill the xylene mixture to drive out more precipitate. At some point, usually after about 3 gassings, the stuff in the xylene starts getting a yellow tinge and then starts to stink like HCl (maybe water in the xylene?) Anyway, I did it in three passes, and as soon as I saw a tiny amount of yellow, I quit. You can use the slightly yellowed stuff -- wash it with acetone, but the more yellow and the more stink, the more of the real product seems to disappear and be dissolved in the water.
When you get the final stuff -- MDMA mixed with xylene -- get as much xylene as you can out of it on a vacuum filter (returning the xylene, possibly, for another passing of the HCl gas). Then, over the vacuum, put in a few jolts of acetone. This dissolves not only the yellow crap (if there is any), but tends to pull out the xylene and things dry quite rapidly.
I got (on the time I made the fewest errors) about 18 grams of MDMA (dried from the acetone) from 25 ml of ketone.
I did not recrystallize this "raw product", since it looked snow-white to me. It does have a faint odor (safrole?) but very faint.
Note by Rhodium: Even if your product looks pure and snow-white to you, please do yourself and your friends a favor and recrystallize the product. A lot of unhealthy byproducts are white/colorless just like pure MDMA, so visual inspection cannot be used to verify purity (you can only verify the presence of colored impurities). Pure MDMA·HCl is odorless, so using your nose is a better way to gauge purity.
The bioassay was extremely successful (I tested perhaps 120mg of acetone-washed product on a human subject weighing about 170 pounds).
Making Doses:
This was a pain. What I finally wound up doing was to take the pile of dried stuff, chopped it with a razor, crushed it with a piece of shiny plastic, chopped again, and got pretty good dust. Then I packed the dust in the most consistent repeatable possible way into capsules. Then I counted the capsules, weighed the whole mess, then weighed that many empty capsules. Thus I determined the dose for each, and wrote that down with the capsules.
Although it's obviously an approximation, if I know that the average capsule in a batch contains 270mg of MDMA, I can pour out that capsule on a sheet of glass, chop it with a razor, and form the powder into a fairly uniform rectangle. Then it's easy to cut out roughly what I want. If you divide the 270mg into thirds, you know that each is about 90mg, et cetera.
A method to measure a known quantity is with an empty capsule and a cylindrical stick or something to pack the stuff solidly into the bottom. I used a drill index and was able to find a perfect sized drill bit for my capsules. Then you can measure the height of the stuff in the capsule pretty accurately.
I made a big error the first time I was loading a bunch of powder into capsules. It took a fair amount of time, and I took a bunch of breaks. I couldn't bear to wash that beautiful white powder off my fingers each time, so I licked them clean. I have no idea what my final dose was that day, but I know it was higher than anything I've taken before or since. The next time I was fairly anal about doing the whole job all at once, followed by a single final lick, (and then 120mg from a capsule, just to make sure).

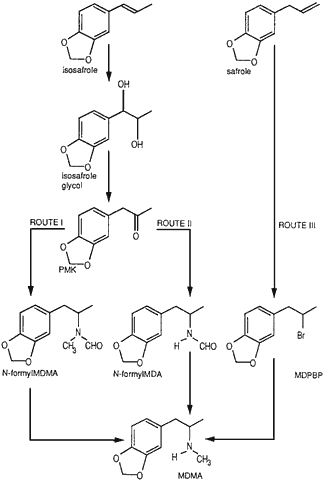 Fig. 1.
Fig. 1.
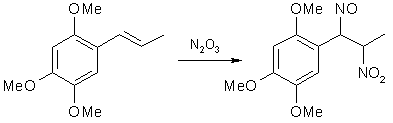 Fig 1. Formation of Asarone Pseudonitrosite
Fig 1. Formation of Asarone Pseudonitrosite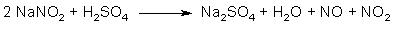 Fig 2. N2O3 from NaNO2 and H2SO4
Fig 2. N2O3 from NaNO2 and H2SO4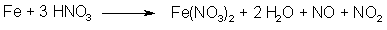 Fig 3. N2O3 from HNO3 and iron
Fig 3. N2O3 from HNO3 and iron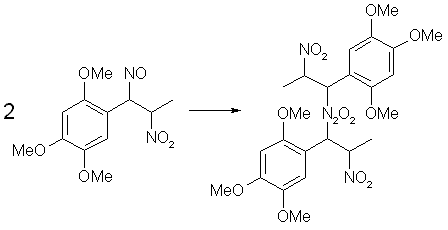 Fig 4. Dimerization of Asarone Pseudonitrosite
Fig 4. Dimerization of Asarone Pseudonitrosite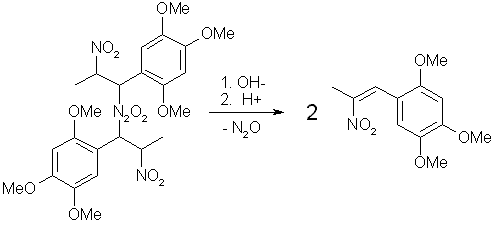 Fig 5. Hydrolysis of Asarone Pseudonitrosite Dimer
Fig 5. Hydrolysis of Asarone Pseudonitrosite Dimer

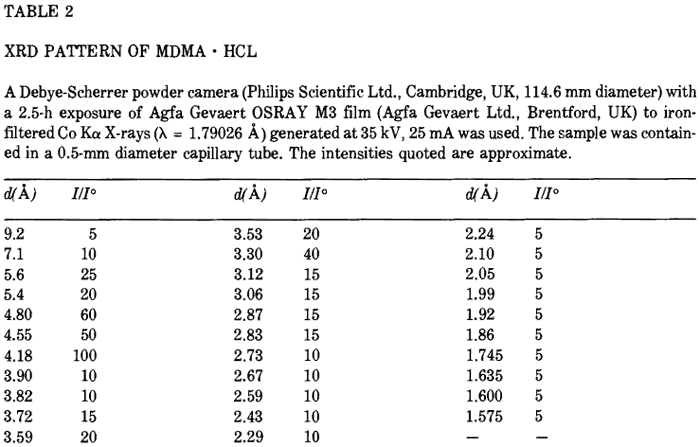{kind=link}
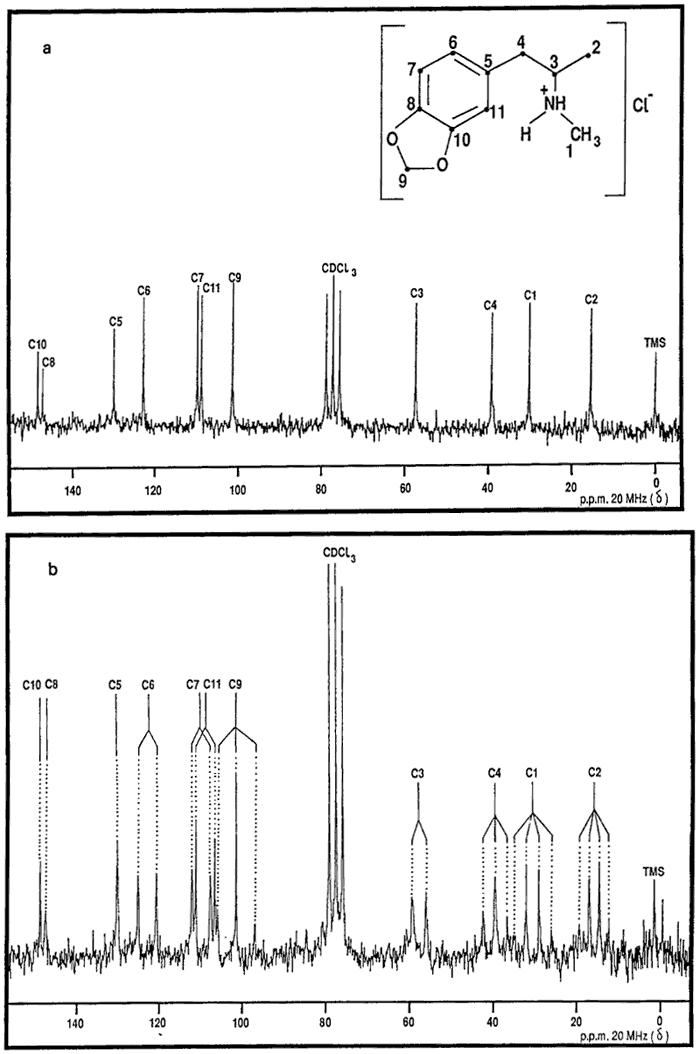{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}